The team investigates the molecular mechanisms driving neurodegeneration in amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), Alzheimer’s disease and in repeat expansion diseases including Huntington’s disease and X-linked Dystonia Parkinsonism (XDP). We use innovative preclinical models and functional genomics approaches to identify new therapeutic targets for neurodegenerative diseases. We have established collaborations with academic and pharmaceutical partners to develop novel approaches to therapy, including RNA-targeting antisense oligonucleotides and immunotherapies for ALS and FTD.
What’s happening?






Publications
Accurate strand-specific long-read transcript isoform discovery and quantification at bulk, single-cell, and single-nucleus resolution
National Library of Medicine (2026)https://www.biorxiv.org/content/10.64898/2026.02.12.705617v1
Recent advances in long-read transcriptome sequencing enable high-throughput profiling of full-length RNA isoforms in bulk, single-cell, and single-nucleus samples. However, long-read datasets typically contain a mixture of complete and partial transcripts, leading to pervasive ambiguity in read-to-isoform assignment and complicating accurate isoform identification and quantification, particularly in the absence of reliable reference annotations.
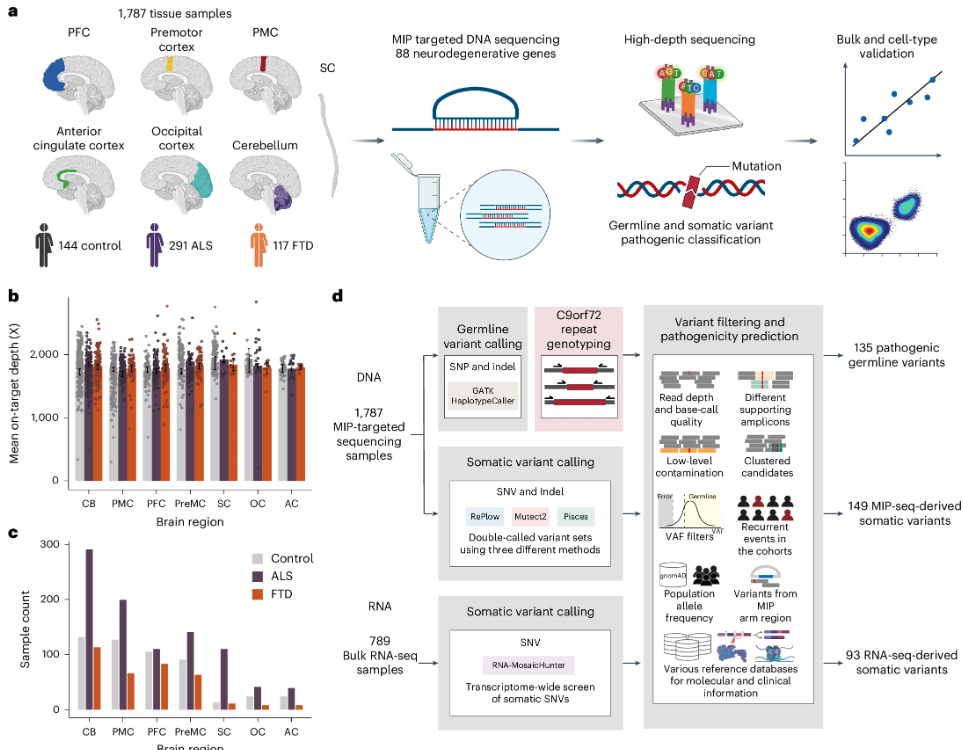
Somatic mosaicism in ALS and FTD identifies focal mutations associated with widespread degeneration
Nature Genetics (2026) https://doi.org/10.1038/s41588-026-02570-6
Although mutations in many genes cause familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), most cases are sporadic (sALS and sFTD) with unclear etiology. Here we tested whether somatic mutations contribute to sALS and sFTD by deep targeted sequencing of 88 neurodegeneration-related genes in postmortem brain and spinal cord samples from 399 sporadic cases and 144 controls.
Statins and genetic inhibition of the mevalonate pathway activate an ATF3-STMN2 regenerative program
Biorxiv (2026) https://doi.org/10.64898/2026.02.23.707492
Loss of neuronal regenerative capacity is a common feature of neurodegenerative disease and axonal injury, yet the transcriptional programs governing this state remain poorly defined. Stathmin-2 (STMN2), a tubulin-binding protein essential for axon maintenance and repair, is profoundly depleted following loss of nuclear TDP-43 in neurodegenerative disease. Here, we identify statins as potent inducers of STMN2 expression.
Impaired nucleocytoplasmic transport in SOD1-mediated ALS
Molecular Neurodegeneration (2026): https://doi.org/10.1186/s13024-026-00930-8
Impaired nucleocytoplasmic transport (NCT) has emerged as a shared pathogenic mechanism in various neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS). Although mutations in the gene encoding superoxide dismutase 1 (SOD1) account for approximately 20% of familial ALS cases, the impact of mutant SOD1 accumulation on the NCT remains unclear.